
Periodic Safety Update Reports (PSURs) are critical documents for medical device and in vitro diagnostic product manufacturers. These reports are a key component of a manufacturer’s post-market surveillance and risk management program, providing ongoing assessment of the safety and performance of their products. In this blog, we will explore what PSURs are, what information they contain, and why they are important. We will also discuss the regulatory requirements for PSURs, the process for preparing and submitting them, and some best practices for ensuring their accuracy and completeness.

Table of Contents
What is a PSUR?
A PSUR is a comprehensive report that provides an evaluation of the safety and performance of a medical device or in vitro diagnostic product over a defined period of time. The report summarizes the manufacturer’s post-market surveillance activities, including adverse event reporting, complaint handling, and other sources of safety information. The purpose of the report is to identify any potential safety concerns associated with the use of the product and to assess the risk-benefit balance.
What information is included in a PSUR?
PSURs typically include the following information:
- Executive Summary: This section provides a brief overview of the report’s content and findings.
- Introduction: This section provides background information on the product, including its intended use, regulatory status, and any significant changes since the previous PSUR.
- Pharmacovigilance Activities: This section describes the manufacturer’s post-market surveillance activities, including adverse event reporting, complaint handling, and other sources of safety information.
- Safety Evaluation: This section summarizes the safety data collected during the reporting period, including any adverse events, trends, and emerging safety concerns.
- Benefit-Risk Assessment: This section evaluates the product’s risk-benefit balance based on the safety and effectiveness data collected during the reporting period.
- Conclusion and Recommendations: This section provides a summary of the report’s findings and any recommendations for further action.
Regulatory Requirements for PSURs:
The regulatory requirements for PSURs vary depending on the jurisdiction and the type of product. In the European Union (EU), PSURs are required for all medical devices and in vitro diagnostic products that have received a CE mark. The frequency of PSUR submission depends on the classification of the product, with higher-risk products requiring more frequent reporting.
In the United States, PSURs are not required by the Food and Drug Administration (FDA) for medical devices or in vitro diagnostic products. However, manufacturers are still required to conduct post-market surveillance activities and report adverse events to the FDA.
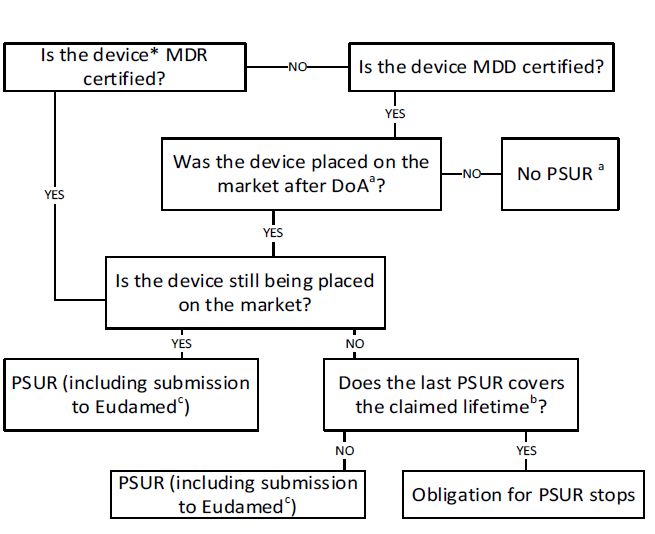
Process for Preparing and Submitting PSURs:
The process for preparing and submitting PSURs involves several steps, including:
- Data Collection: The manufacturer collects safety and performance data for the reporting period, including adverse events, complaint handling, and other sources of safety information.
- Data Analysis: The manufacturer analyses the data to identify any safety concerns or emerging trends.
- Report Writing: The manufacturer prepares the PSUR, including an executive summary, introduction, safety evaluation, benefit-risk assessment, and conclusions.
- Internal Review: The manufacturer conducts an internal review of the PSUR to ensure its accuracy and completeness.
- Submission: The manufacturer submits the PSUR to the regulatory authority or notified body, as required.
Workflow for assessment of PSUR requirement:
To ensure the accuracy and completeness of PSURs, manufacturers should follow some best practices, including:
- Establishing a robust post-market surveillance program that includes the collection and analysis of safety and performance data.
- Conducting regular internal reviews of safety data to identify emerging safety concerns.
- Ensuring that all adverse events and complaints are reported in a timely manner to the regulatory authority or notified body.
- Designing PSURs to be comprehensive and transparent, with clear and concise
- Involving cross-functional teams in the preparation of PSURs, including representatives from regulatory affairs, clinical development, and quality assurance.
- Providing a clear and detailed description of the product and its intended use, as well as any significant changes since the previous PSUR.
- Ensuring that the safety evaluation and benefit-risk assessment sections are based on robust data and analysis and that any limitations or uncertainties are clearly communicated.
- Providing a comprehensive summary of adverse events and other safety data, including a description of the severity, frequency, and outcome of each event.
- Including a thorough review of the product’s labeling and instructions for use, to ensure that they are accurate and up to date.
- Continuously monitoring the safety and performance of the product and updating the PSUR as needed.
How Can RegDesk Help?
RegDesk is a holistic Regulatory Information Management System that provides medical device and pharma companies with regulatory intelligence for over 120 markets worldwide. It can help you prepare and publish global applications, manage standards, run change assessments, and obtain real-time alerts on regulatory changes through a centralized platform. Our clients also have access to our network of over 4000 compliance experts worldwide to obtain verification on critical questions. Global expansion has never been this simple.




