
This article was updated June 2, 2025. Ireland’s medical device regulations are harmonized with the European Union’s Medical Devices Regulation (MDR) and In Vitro Diagnostic Medical Devices Regulation (IVDR). The Health Products Regulatory Authority (HPRA) is the national competent authority responsible for regulating medical devices in Ireland.

Overview
To market a medical device in Ireland, manufacturers must first obtain CE marking, demonstrating conformity with EU regulations. Once CE marked and registered in one EU country, the device can be marketed throughout the EU. HPRA oversees medical devices and other health products, including cosmetics, clinical trials, tissues and cells, blood components, controlled drugs, and veterinary medicines.
Device Classification:
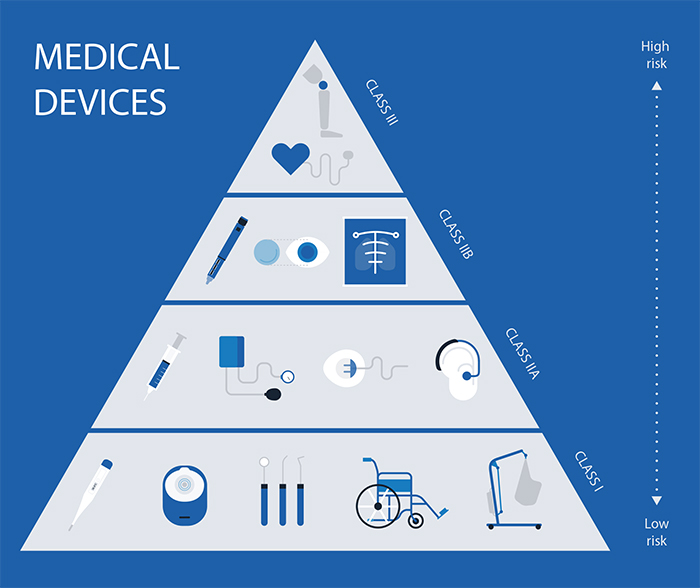
Devices are classified according to EU rules into:
Class I
Class lla
Class Ilb
Class III
Higher classes correspond to higher risk and more stringent regulatory scrutiny.
Ireland Medical device registration Requirements
CE Marking: All devices must be CE marked before being placed on the Irish market.
EUDAMED Registration: The EU’s EUDAMED database is central to MDR/IVDR compliance. Economic operators (manufacturers, authorized representatives, importers) must register in EUDAMED. The “Actors” module is active, and other modules are being phased in.
HPRA National Registration: In addition to EUDAMED, certain economic operators in Ireland must register with HPRA:
Distributors and manufacturers based in Ireland not covered by EUDAMED registration must register domestically with HPRA.
Health institutions (e.g., hospitals, clinics) manufacturing devices must also register with HPRA.
Custom-made device manufacturers (Class I, IIa, IIb) register with HPRA; custom-made Class III/implantable devices register with EUDAMED.
Device Registration: In the absence of the EUDAMED device registration module, MDR/IVDR-compliant Class I devices, system/procedure packs, custom-made devices, and IVDs should be registered with HPRA using official MDR/IVDR spreadsheets. Legacy devices (certified under old Directives) can be voluntarily registered using the MDR Legacy Spreadsheet. Legacy devices may be marketed until May 27, 2024, and made available until May 27, 2025.
Registration Numbers: Economic operators should ideally have a Single Registration Number (SRN) from EUDAMED or a HPRA registration number before registering devices.
Authorized Representative:
Non-EU manufacturers must appoint an Authorized Representative (AR) in the EU to assist with registration and compliance.
Quality Management System:
A Quality Management System (QMS), such as ISO 13485 certification, is recommended. ISO 13485 certificates are typically valid for three years, while EU device certificates are valid for five
years.
Timeframes:
-Class I devices: Registration typically takes 4-6 weeks.
-Class III devices: Registration can take several months due to higher risk and scrutiny.
Wholesale Distribution Authorizations (WDA):
Entities distributing healthcare products must apply for a WDA from HPRA. Requirements include:
Permanent physical site in Ireland for wholesale activities.
Details of all sites and relevant authorizations.
Technical agreements and evidence of Good Distribution Practice compliance. Audit completion and any special product authorizations.
Recent Updates:
HPRA has streamlined the Certificate of Free Sale process with a unified application form, new Excel device schedule, updated certificate template, and a dedicated email for submissions.
HPRA is developing a new medical device portal for registrations, which will eventually synchronize with EUDAMED to avoid dual data entry. No launch date has been announced.
Summary: Key Steps for Medical Device Registration in Ireland
CE Marking
EUDAMED Registration
HPRA National Registration
Device Registration
Appoint Authorized Rep.
QMS Certification
WDA (if disturbing)
Requirement/Action:
Obtain CE mark for device compliance with EU MDR/IVDR
Register as an economic operator (mandatory for most actors)
Register with HPRA if required (distributors, manufacturers, health institutions)
Submit device details to HPRA (if EUDAMED module unavailable)
Non-EU manufacturers must appoint an EU Authorized Representative
Implement ISO 13485 or equivalent QMS
Apply for Wholesale Distribution Authorization if distributing devices
