
What Is It?
ISO 13485 is a quality management system that is used internationally. It controls safety in work environments, risk management, and design, and creates requirements for inspections, documents, procedures, and verification of safety and effectiveness of medical devices.
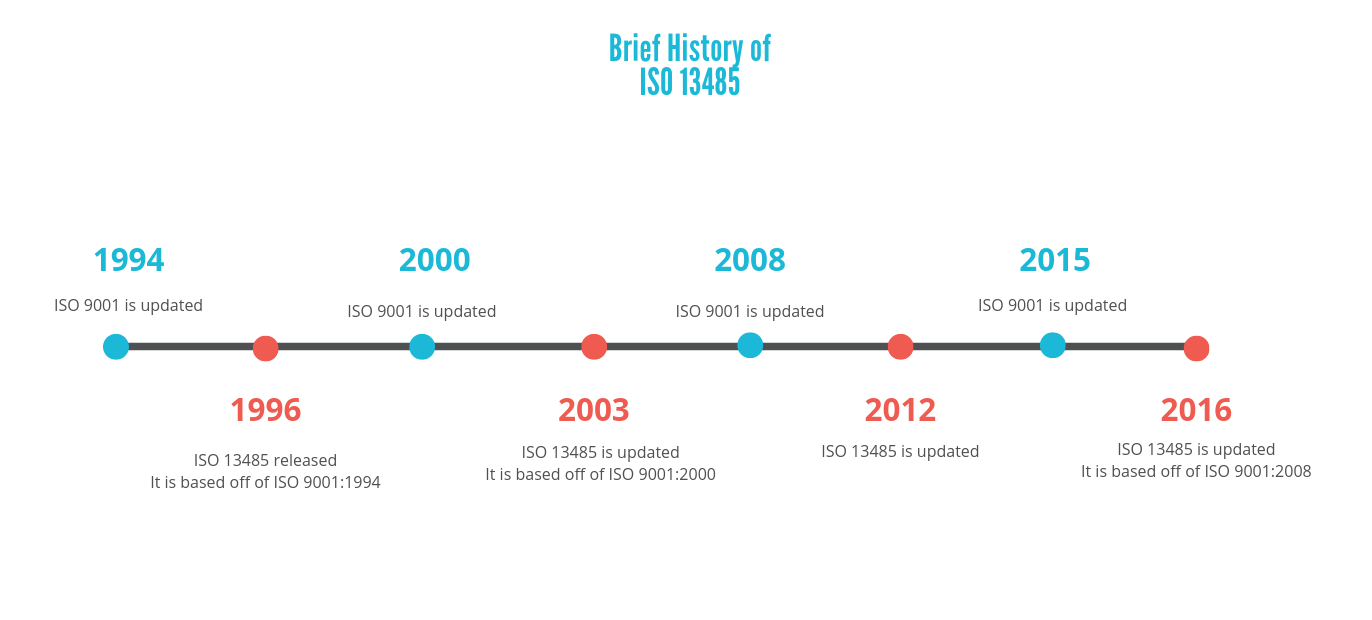
There is a pattern presented here, where the updates of these quality management systems generally alternate, and the ISO 13485 updates resemble the latest versions of ISO 9001s. However, although ISO 13485 is similar to the other system, does not mean that it replaces it. A manufacturer would still have to meet the requirements presented in ISO 9001 if they want to follow this system.
Changes Between ISO 13485 Versions
The most current ISO 13485 version that is used is from 2016. Here is a comparison of the latest revision to the 2012 version:

In this new version, ISO 13485 better covers the requirements needed to export to the U.S. market, which is in 21 CFR part 820, so manufacturers following the ISO should have an easier time expanding. The newer version has also added in a lot more requirements and standards for manufacturers to follow that were not present in previous versions of ISO.
How RegDesk Can Help
It is important to understand quality and safety requirements whether a company intends to distribute products in the U.S. or elsewhere. This shift in the ISO quality requirements will bring in new changes that are important for manufacturers to know about. RegDesk provides medical device companies with all the safety requirements and regulation information required to help them register their devices and distribute them to the market. We track and monitor any ISO changes in order to give our clients the most up-to-date information. RegDesk also provides information for over 100 markets worldwide to help medical device and pharmaceutical companies expand internationally.
