
The Food and Drug Administration (FDA) recently approved “Ocaliva”, a drug for the treatment of a chronic and rare liver disease. As Ocaliva indicated significant clinical benefits as compared to its only pre-existing therapy, Ursodeoxycholic acid (UDCA), its development and approval were expedited. Increasing the rate of approval for drugs that treat serious diseases where medical needs are unmet or inadequately done is essential to the health and safety of patients. There are four different approaches to making drug access as rapid as possible: fast track designation, accelerated approval program, breakthrough therapy and priority review. In regards to rare diseases, orphan drug designation is also a noteworthy aspect to consider in conjunction with the four other processes. For example, Ocaliva was granted the FDA’s fast track designation, accelerated approval program, and orphan drug designation. So what exactly are these processes and their distinctions?
They are all similar in their core function. That is, they serve to get important new drugs to patients more rapidly. The drug must significantly aid in the treatment, prevention or cure of a critical condition. A “serious condition” is judged to be severely detrimental to the day-to-day functioning and affect survival. The drug addresses an unmet medical need whether there is no drug, or the available drug is medically inadequate. Additionally, drugs will usually qualify for more than one program which can further accelerate its accessibility.
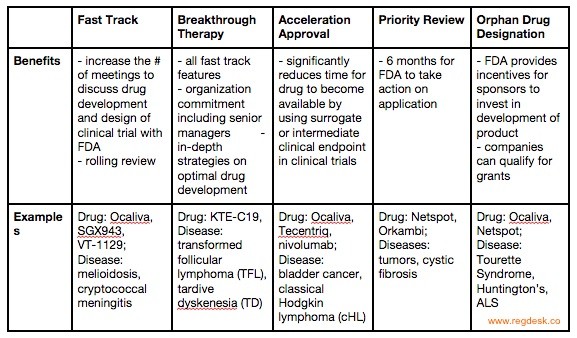
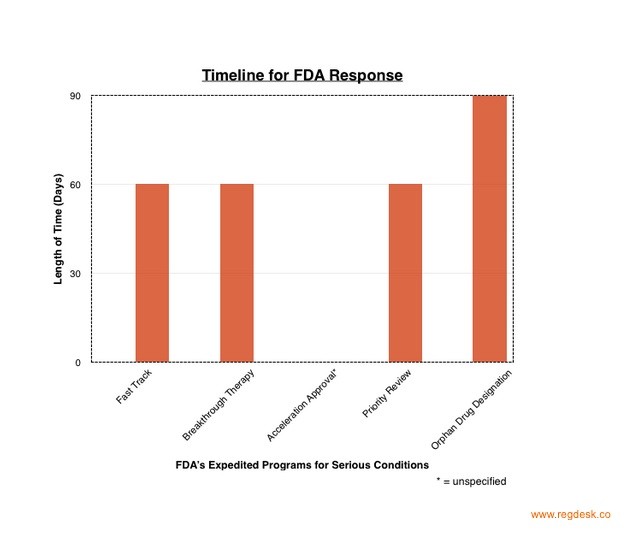
The Fast Track Designation ensures that the FDA takes action to accelerate the development and review of a drug. This acceleration is achieved by increasing the number of meetings companies have with the FDA to discuss the drug’s development and the design of clinical trials for data collection. While standard reviews require the application of a complete New Drug Application (NDA), the Fast Track allows a rolling review. A rolling review allows clients to provide sections of their NDA application as they complete them. The drug company can request Fast Track designation at any time. The FDA will respond to the request within 60 days. Eligibility for the Fast Track also allows Eligibility for Accelerated Approval and Priority Review, if relevant.
Breakthrough Therapy Designation applies to drugs that show a significant advantage over available therapy in preliminary clinical trials. The treatment is measured by a clinically significant endpoint that measures the drug’s effect on irreversibility morbidity or mortality (IMM) or on severe symptoms that occur as a consequence of the disease. Drugs granted the Breakthrough Therapy designation are given all Fast Track features, organization commitment from senior managers and in-depth strategies on optimal drug development. The pharmaceutical company can request the Breakthrough Therapy Designation or be suggested by the FDA, usually no later than the end-of-phase-2 meetings. The FDA responds within 60 days of receiving the request.
Accelerated Approval is an expedited pathway for qualified drugs to be approved according to data collected in clinical trials using a surrogate endpoint. Congress approved the use of surrogate or an intermediate clinical endpoint to accelerate drug approval for serious diseases with unmet medical needs in 2012 under the FDA Safety Innovations Act. The surrogate endpoint is a marker whose measurement can predict clinical benefits, though it is not a direct measure of clinical benefits. While phase 4 confirmatory trials must continue to be conducted after successful drug approval through Accelerated Approval Pathway, this significantly reduces the time in which a drug becomes available.
All drugs marketed in the US must undergo a thorough FDA review before approval. The FDA’s Prescription Drug User Act (PDUFA) passed in 1992 in response to a clear distinction between the urgency in which various drugs should be made accessible. The PDUFA segregated drug reviewal into two systems: Standard Review and Priority Review. The Priority Review ensures that the FDA takes action on an application within six months, rather than the ten months allowed for Standard Review. Drugs that can treat, diagnose or prevent serious diseases that are currently medically unmet, can qualify for Priority Review. Companies must pay a fee which the FDA will use to support and expand its resources. In return, the FDA establishes deadlines for completing the review of the company’s drug application. The drug must show significantly increased effectiveness and efficiency. Companies can apply for Priority Review and expect an FDA response within 60 days. Being granted Priority Review does not change the scientific and medical standards to which it is held for approval; for example, clinical trial periods will remain the same length.
Orphan Production Development
While Orphan Production Development is not one of the four approaches listed under the FDA site as a method for accelerating drug accessibility, it is often used in conjunction with relevant drugs. Relevant drugs are those that diagnose or treat rare diseases or conditions whose medical needs are currently unmet. If designated “Orphan Status”, the drug undergoes an advanced evaluation. As “orphan drugs” address the needs of patients with rare conditions, they are produced with the expectation of small sales and financial loss. The FDA uses incentives to encourage sponsors to invest in developing the product. Additionally, companies with orphan drugs can qualify for grants that aid in the funding for clinical research. Diseases such as Huntington’s disease, ALS, Tourette syndrome all affect a small subset of patients in the US and are thus considered “rare” by the FDA. Drugs treating these diseases can qualify as orphan drugs. Companies should apply for orphan status in early development, and can expect less than 90 days for the FDA’s response.
This article has provided a very basic overview of the five possible approaches to increasing the rate of drug approval. For further details, please visit the FDA site. RegDesk can help companies seeking in-depth guidance on how regulations may or may not apply to their particular products. With access to a network of leading experts, RegDesk can help Life Sciences companies form strategies for the optimal approach. If you have any queries, please visit our site.
