
The Food and Drug Administration (FDA or the Agency), the US regulating authority in the sphere of medical devices and other healthcare products, has published a guidance document dedicated to deciding when to submit a 510(k) for a software change to an existing device.
The latest version of the document was issued in October 2017. Apart from a general overview of the principles to be applied, the document describes in detail how such a decision should be taken by a responsible party. It is also important to mention that due to its legal nature, the guidance does not introduce additional requirements itself but provides clarifications and recommendations to be considered by all the parties involved in order to ensure and sustain compliance with the applicable regulatory requirements. Moreover, an alternative approach could be applied, provided it complies with the current legislation and has been approved by the FDA in advance.

Table of Contents
In particular, the guidance describes how the responsible party shall decide whether the software change to a medical device already placed on the market should be treated as significant and thus require a new 510(k) premarket notification to be duly submitted. The approach described in the document is considered to be the least burdensome for all the parties involved in operations with medical devices. The document also contains references to the FDA-recognized voluntary consensus standards medical device manufacturers may refer to when demonstrating compliance with the applicable regulatory requirements in terms of safety and performance.
Applying the Approach
In order to assist medical device manufacturers in applying the main principles outlined therein, the document provides a flowchart accompanied with additional clarifications and examples intended to guide the responsible party through the process of making a decision regarding the necessity of a new premarket notification 510(k) related to a software change made to a medical device already approved for marketing and use in the US. It is stated that the scheme provided cannot cover all possible intricacies related to such changes and how they may affect the decision, so it describes only the general approach suggested. Hence, when determining the necessity of a new premarket notification 510(k), the medical device manufacturers shall consider both general principles and the flowchart provided in the guidance, as well as additional factors highlighted in a separate section of the document.
The approach described in the flowchart is designed to answer specific questions regarding a software change made to an existing medical device. It is intended to guide the manufacturer in determining whether a new submission is required or not based on the nature of the changes made. Under the general rule, the manufacturer shall make its decision based on a comparison to the initial medical device approved to be marketed and used and evaluate how the software changes will impact its safety and performance. According to the guidance, manufacturers are required to submit a new 510(k) when a change (or changes) exceed the 21 CFR 807.81(a)(3) threshold, e.g., it “could significantly affect the safety or effectiveness of the device,” or constitutes a “major change or modification in the intended use of the device.” The Agency emphasizes that in some cases, a single change could constitute numerous changes that could cause a positive or negative effect. In order to assist with applying this approach, the FDA also provides several examples of justifications (rationale) the medical device manufacturer may use.
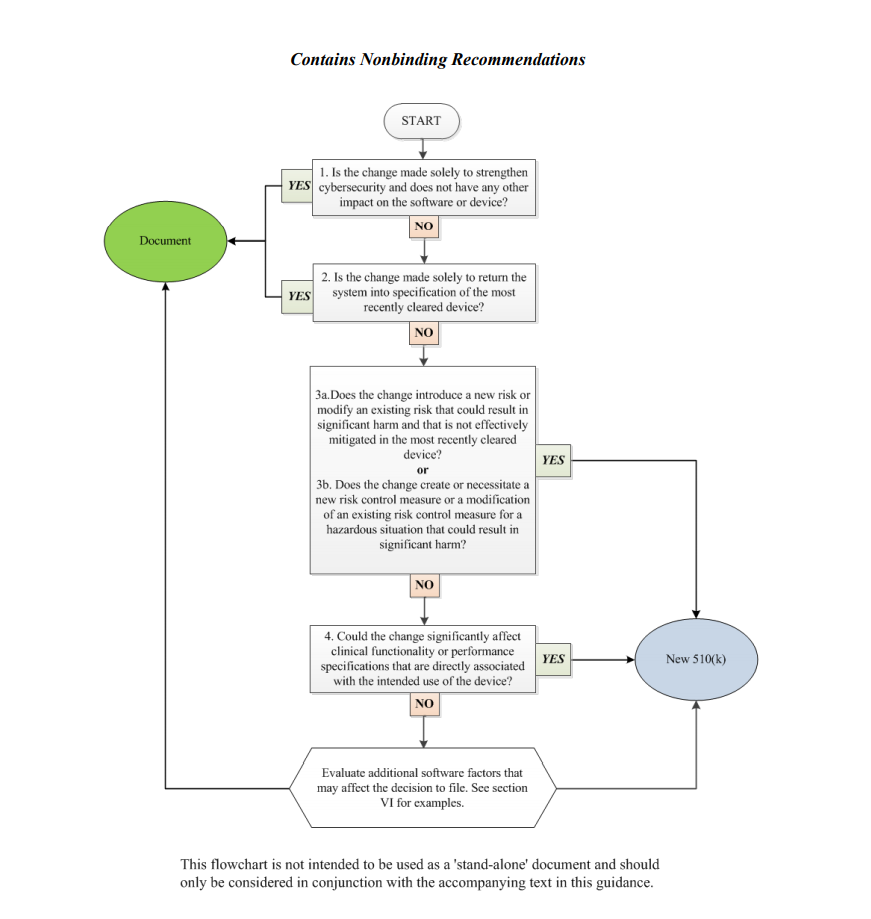
Decision-Making Process: the Flowchart
In the course of the decision-making process described in a flowchart, the medical device manufacturer shall answer the following questions:
1. Is the change made solely to strengthen cybersecurity and does not have any other impact on the software or device?
According to the document, most of the changes intended exclusively to improve cybersecurity do not require a new submission, provided they do not affect the safety and performance of a medical device in general. However, should the manufacturer identify such an impact, it should move forward with answering the rest of the questions provided in a flowchart to make a decision.
2. Is the change made solely to return the system to specification of the most recently cleared device?
In most cases, if the changes are intended solely to restore the medical device in question to the specifications of a medical device already approved for marketing and use, such changes would not require a new submission, as long as such changes do not cause a significant impact to the safety and effectiveness of a medical device, or its intended use. In this regard, the medical device manufacturer shall conduct a rigorous analysis in order to evaluate the impact caused by such changes. It is stated that in the case of missing or ambiguous software requirements, further steps as per the flowchart would be required.
3. Does the change introduce a new risk or modify an existing risk that could result in significant harm and that is not effectively mitigated in the most recently cleared device?
In this regard, the manufacturer shall identify whether the changes made to the software result in certain changes to the risks associated with a medical device. According to the document, if the software change creates new risks, modifies existing ones, makes the potential harm associated with the risks more severe, or if the risks in question are not properly mitigated in an existing device, further consideration would probably be required.
4. Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm?
If the newly introduced risk control measures could potentially impact a medical device’s overall safety and performance, an additional evaluation would be required. Should such control measures be reasonably necessary to address new risks, submission of a new premarket notification 510(k) would be required. At the same time, a new submission is not required if the new risk control measures are intended to improve existing ones, provided that the latter were sufficient to mitigate the risks associated with the device.
5. Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device?
If the software changes made to an existing medical device affect its clinical performance, submission of a new premarket notification 510(k) would be required.
Apart from answering the questions listed above, the medical device manufacturer shall also evaluate additional software factors that may affect the decision to submit a new premarket notification 510(k).
In summary, the present FDA guidance describes in detail the approach to be applied by the medical device manufacturers when deciding whether the software changes to an existing medical device require submission of a new 510(k). For this purpose, the document provides a flowchart describing a step-by-step procedure to be followed in order to make such a decision.
Sources:
How Can RegDesk Help?
RegDesk is a next-generation web-based software for medical device and IVD companies. Our cutting-edge platform uses machine learning to provide regulatory intelligence, application preparation, submission, and approvals management globally. Our clients also have access to our network of over 4000 compliance experts worldwide to obtain verification on critical questions. Applications that normally take 6 months to prepare can now be prepared within 6 days using RegDesk Dash(TM). Global expansion has never been this simple.
