Associated Country
 European Union
European Union Medical devices are an integral part of healthcare, and their safety and efficacy are critical to ensure patient well-being. In 2017, the European Parliament and Council adopted the new Medical Devices Regulation (MDR) and In-Vitro Diagnostic Regulation (IVDR) to replace the existing Medical Devices Directive (MDD) and In-Vitro Diagnostic Directive (IVDD). The new regulations aim to ensure that medical devices and in-vitro diagnostic devices (IVDs) meet the highest standards of safety and performance while improving transparency and accountability throughout the supply chain.
In this blog, we will provide a comprehensive overview of the EU MDR, including its purpose, scope, and key changes. We will also discuss the implications of these changes for medical device manufacturers and the steps they need to take to comply with the new regulation.
Implications of the EU MDR for Medical Device Manufacturers:
The primary objective of the EU MDR is to ensure the safety and effectiveness of medical devices in the EU. The regulation aims to achieve this by improving the transparency and accountability of medical device manufacturers, strengthening clinical evaluation requirements, and enhancing post-market surveillance.
The scope of the EU MDR is broad and covers all medical devices, including in vitro diagnostic medical devices (IVDs). This includes products such as pacemakers, surgical instruments, and diagnostic imaging equipment. The regulation applies to all medical device manufacturers, importers, and distributors that sell their products in the EU.
Key Changes Introduced by the EU MDR:
The EU MDR introduces several significant changes to the medical device industry. Some of the key changes are as follows:
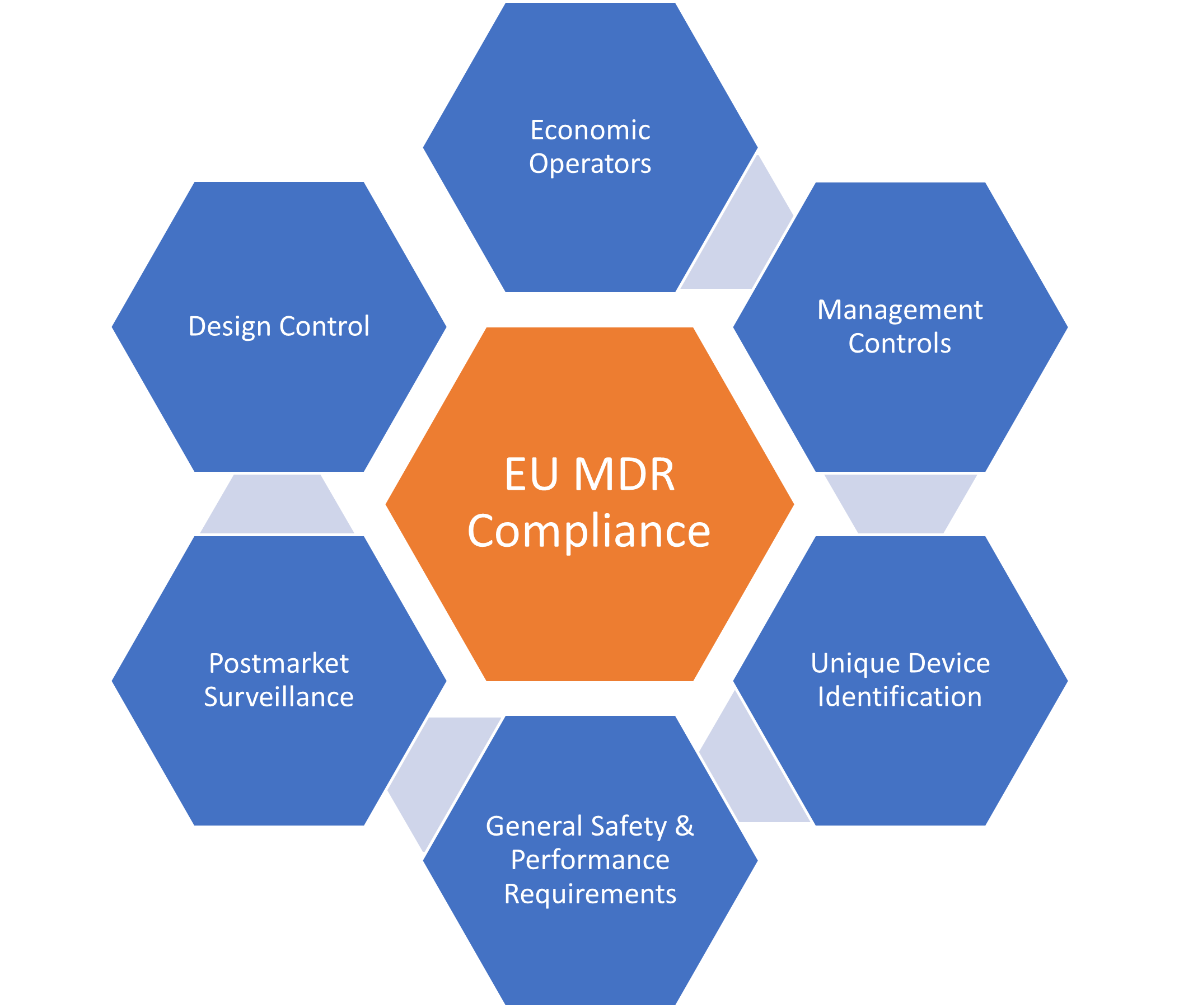
I. Increased Clinical Evidence Requirements: The EU MDR introduces more stringent requirements for clinical evidence. Manufacturers must demonstrate that their products are safe and effective based on clinical data obtained from well-designed clinical trials. This requirement applies to all classes of medical devices, including low-risk devices.
II. Strengthened Post-Market Surveillance: The EU MDR requires manufacturers to monitor the performance of their products in the market continuously. This includes the reporting of adverse events, incidents, and near-misses. The regulation also requires manufacturers to implement a post-market surveillance system to monitor their products’ performance and take appropriate corrective actions when necessary.
III. Enhanced Traceability and Unique Device Identification (UDI): The EU MDR requires all medical devices to carry a unique device identification (UDI) code. This code should be easily readable and should provide information about the device’s identity, manufacturer, and other relevant details. This requirement aims to enhance the traceability of medical devices and improve patient safety.
IV. Stricter Requirements for Notified Bodies: The EU MDR introduces stricter requirements for notified bodies, and the organizations responsible for certifying medical devices. Notified bodies must now demonstrate that they have the necessary expertise and resources to carry out their certification duties effectively.
Implications of the EU MDR for Medical Device Manufacturers:
The EU MDR will have significant implications for medical device manufacturers. Manufacturers must comply with the new requirements by May 26, 2021, to continue selling their products in the EU.
Manufacturers will need to invest in additional resources to meet the increased clinical evidence requirements. They will also need to establish a post-market surveillance system to monitor the performance of their products continually. The enhanced traceability requirements will require manufacturers to implement new labeling systems and processes.
Manufacturers will also need to work closely with their notified bodies to ensure that their products comply with the new requirements. The stricter requirements for notified bodies may result in delays in certification and increased costs for manufacturers.
Roadmap to Comply with the EU MDR
To comply with the EU MDR, medical, The European Union Medical Device Regulation (EU MDR) is a set of guidelines and requirements that medical device manufacturers must comply with to sell their products in the European Union. Here are the steps to comply with the EU MDR for your medical device.
- Determine the Classification of your Medical Device: Classify your device according to its risk level. This will determine the conformity assessment procedure that needs to be applied.
- Identify the Appropriate Conformity Assessment Route: Depending on the classification of your device, you may need to undergo a conformity assessment procedure with a Notified Body. You must select the appropriate conformity assessment route for your device.
- Ensure that Your Technical Documentation is Compliant: The technical documentation of your medical device must be compliant with the EU MDR requirements. You should review your technical documentation and update it as needed to ensure compliance.
- Appoint an Authorized Representative: Manufacturers outside of the EU must appoint an authorized representative in the EU. The authorized representative must ensure that the manufacturer is compliant with the EU MDR requirements.
- Implement Post-Market Surveillance and Vigilance Systems: You must establish post-market surveillance and vigilance systems to monitor the performance of your medical device in the market and report any adverse events.
- Register your Device in the EUDAMED database: You must register your device in the EUDAMED database before placing it on the market.
- Ensure that your Labeling and Packaging are Compliant: Your device labeling and packaging must be compliant with the EU MDR requirements. Ensure that the labeling and packaging contain all required information.
- Establish a Quality Management System: You must establish a quality management system (QMS) that complies with the EU MDR requirements. The QMS should cover all aspects of your medical device’s lifecycle, from design to post-market surveillance.
- Ensure Compliance with Unique Device Identification (UDI) Requirements: You must comply with the EU MDR requirements for unique device identification (UDI). This involves assigning a unique identifier to your device and registering it in the EUDAMED database.
- Ensure Compliance with Clinical Evaluation Requirements: You must conduct a clinical evaluation of your medical device to ensure that it is safe and effective for its intended use. The clinical evaluation should be based on relevant clinical data and follow the requirements of the EU MDR.
- Post-market clinical follow-up (PMCF): One component of PMS activities that can prove the clinical effectiveness and safety of the device and guarantee ongoing risk acceptability is a post-market clinical follow-up (PMCF). PMCFs are far more official and proactive than other PMS activities, with pre-approved outcomes or acceptance criteria. Usually, the focus is on a certain performance or safety area that is determined by risk management, CER, or even other PMS data. When there are long-term data gaps or unresolved questions related to the use, specific indications, or novel characteristics of a new device, a PMCF is frequently necessary where there is little clinical data for a legacy device.
A PMCF could be a planned follow-up with patients who have been using the device, a clinical study, a suitable registry, a consumer survey, input from key opinion leaders, etc. Understanding the best strategy is a major difficulty for manufacturers because of the variety of PMCF types and the requirement to customize each one uniquely for each device.
By following these steps, you can ensure that your medical device complies with the EU MDR requirements and is ready for sale in the European Union.
Recent Update
EU MDR transitional period and deletion of the MDR/IVDR ‘sell-off dates’ officially implemented on March 20th, 2023.
Regulation (EU) 2023/607 of the European Parliament and of the Council of March 15, 2023, amending Regulations (EU) 2017/745 and (EU) 2017/746, has been published.
The European Union has set deadlines for the certification and sale of various medical devices. The deadline for custom-made Class III implantable devices is May 26, 2026. Class III and Class IIb implantable devices must be certified by December 31, 2027, while Class IIb non-implantable devices must be certified by December 31, 2028. The deadline for Class IIa and Class I (Is/Im) devices is also December 31, 2028. Certificates issued before May 26, 2021, will remain valid.
Sources
Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0746
European Union. (2023). Regulation (EU) 2023/607 of the European Parliament and of the Council of 21 March 2023 https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32023R0607
How Can RegDesk Help?
RegDesk is an AI-powered Regulatory Information Management System (RIMS) designed to simplify global compliance for medical device companies. With regulatory intelligence covering 120+ markets, RegDesk helps you prepare and publish global submissions, manage standards, conduct impact assessments, and stay ahead of regulatory changes all from a single, centralized platform. Expanding into new markets has never been easier.
Associated Country
European Union Medical devices are an integral part of healthcare, and their safety and efficacy are critical to ensure patient well-being. In 2017, the European Parliament and Council adopted the new Medical Devices Regulation (MDR) and In-Vitro Diagnostic Regulation (IVDR) to replace the existing Medical Devices Directive (MDD) and In-Vitro Diagnostic Directive (IVDD). The new regulations aim to ensure that medical devices and in-vitro diagnostic devices (IVDs) meet the highest standards of safety and performance while improving transparency and accountability throughout the supply chain.
In this blog, we will provide a comprehensive overview of the EU MDR, including its purpose, scope, and key changes. We will also discuss the implications of these changes for medical device manufacturers and the steps they need to take to comply with the new regulation.