Medical Device AI Submission Generator
Submission-Ready Applications in Days with AI
Registering a medical device is fragmented and redundant, so often, the process is lengthy and overly complicated. Delays can cost millions of dollars, and most companies fail to meet their annual product submission goals.
RegDesk leverages the Power of AI to help you get instant access to country-specific templates and auto-fill subsequent applications. That means you can prepare submission-ready applications in days rather than months.
We process several hundred international applications per year. We would always experience delays. With RegDesk our team is able to generate applications within a few hours rather than months.
Chris S., Regulatory Affairs Manager,
Medium-Sized Medical Device Company
WITH REGDESK'S MEDICAL DEVICE AI SUBMISSION
GENERATOR, YOU CAN:
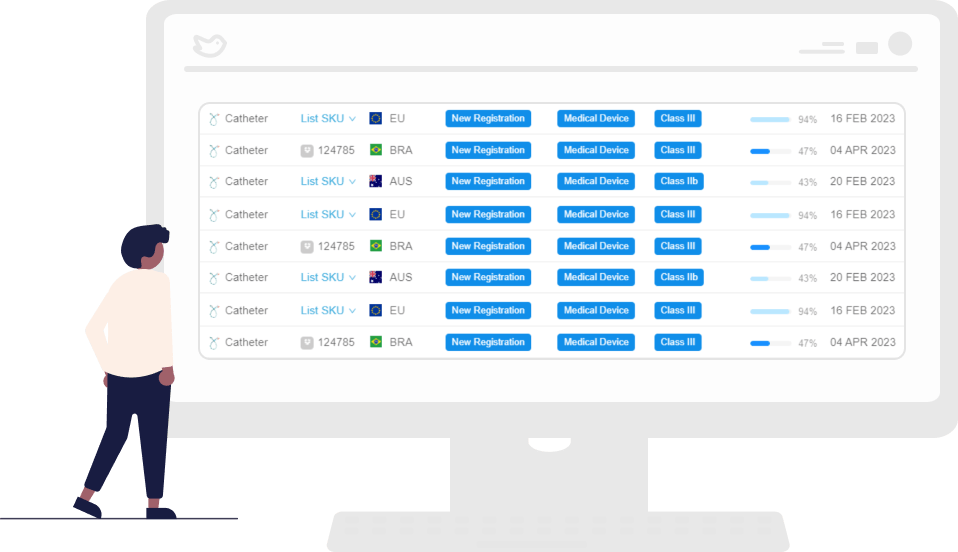
Access Country-Specific Templates Instantly
Streamline the creation and management of international submissions with RegDesk’s comprehensive, country-specific medical device registration templates. Simply select the relevant country template and quickly and accurately build your application in accordance with local requirements.

Prepare Submission-Ready Applications Efficiently
RegDesk’s cloud-based software uses AI to help you prepare submission-ready applications in weeks versus months. Our intuitive software collects, and assembles necessary data, documents and evidence required for medical device submissions.
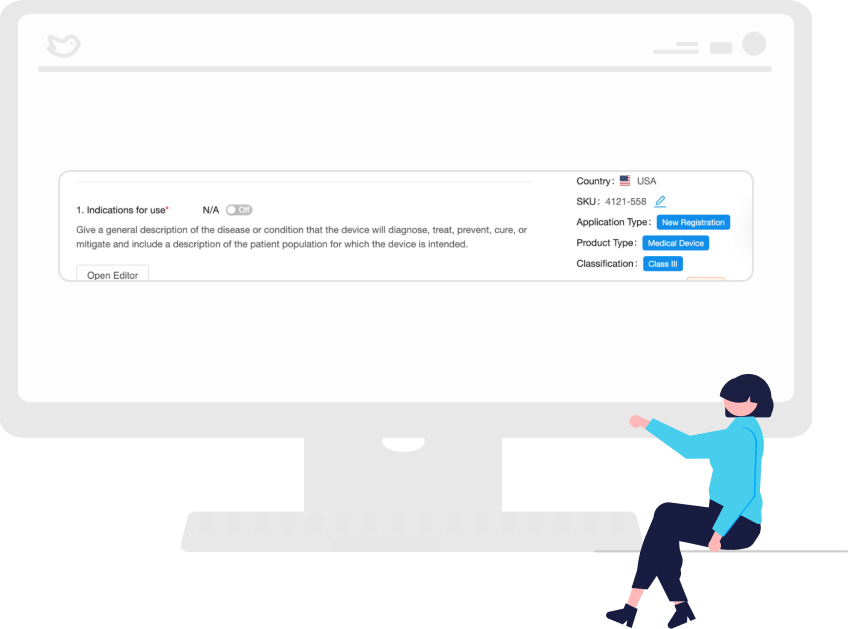
Autofill Subsequent Applications Using AI Technology
Why should you have to keep providing the same information over and over? With RegDesk, you won’t have to.
With our AI, your work on previous applications is used to auto-complete subsequent submissions, thus saving your team time and resources while reducing the risk of non-compliance.
Autofill Subsequent Applications Using AI Technology
With our AI, your work on previous applications is used to auto-complete subsequent submissions, thus saving your team time and resources while reducing the risk of non-compliance.
Medical Device Registration and Publishing Software
RegDesk’s AI Submission Generator simplifies the submission process with simple, fillable forms and helps you generate submission-ready applications within one week instead of three to six months.
Our platform is tailored to medical device companies to help produce jurisdiction-specific submission dossiers and autofill subsequent applications for the same product in different countries within seconds. For instance, if you’ve completed the FDA medical device approval process, you can auto-transfer much of the same information when applying to other countries to help save you time.
Our medical device registration and publishing tool incorporates the most up-to-date requirements to reduce the number of deficiencies you receive from Health Agencies. It has auto formatting, which allows you to prepare submission ready applications.
When you can submit your application quicker, you can receive medical device approval faster, which means more revenue for your company.
Medical Device Registration Process Timeline
On average, it takes regulatory teams between four to six months to prepare, format, and publish a single product submission for a single country. Most mid to large medical device companies have an annual quote to submit several hundred global applications annually.
When it comes to submission generation and publishing, regulatory teams currently:

Waste time on redundant activities such as copying and pasting similar data for multiple submissions.

Spend hours compiling documents for global distributors and corresponding with them.

Have a lack of knowledge of regulatory requirements and formats for international markets.

Feel overwhelmed with the amount of work required to prepare and manage global submissions.

Expend hours of their time formatting submissions.
The current medical device registration process is manual, tedious, and prone to human error. The right medical device registration and publishing software can streamline the process by eliminating redundancies and mistakes to help get your medical device approval faster.
