Table of Contents
How To Bring Medical Devices To Market?
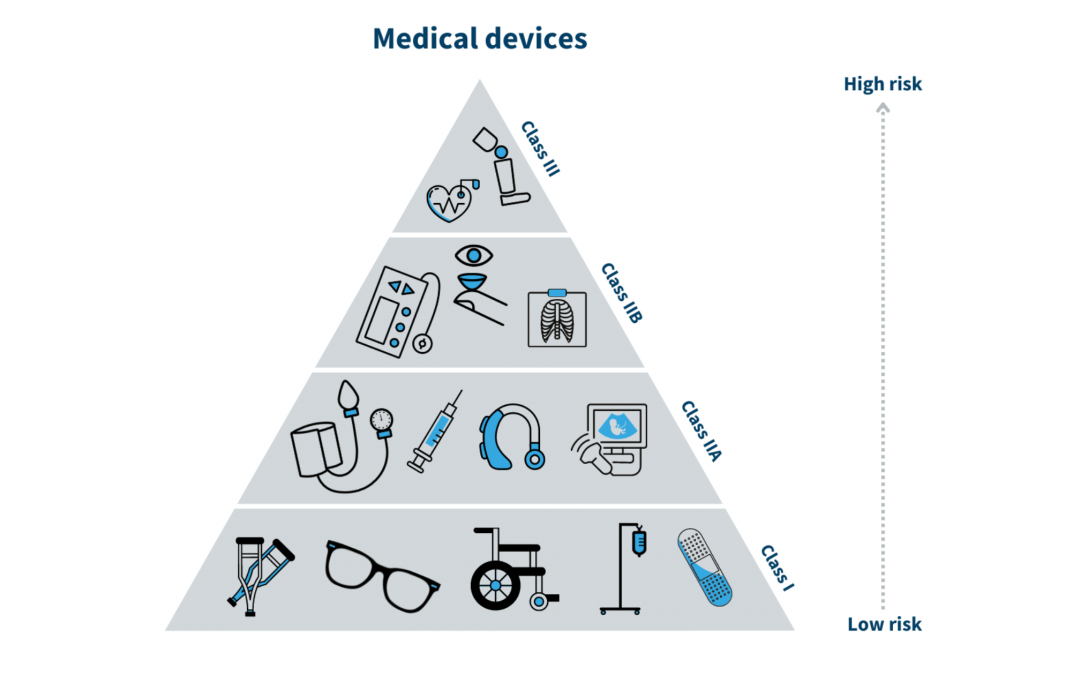
The Food and Drug Administration (FDA) is a federal agency in the United States that is responsible for regulating medical devices, as well as drugs, biologics, and other health-related products. The FDA classifies medical devices into three categories based on their potential for harm:
The classification of a device determines the level of regulation and oversight that it will be subject to. The European Commission is the equivalent agency for the European Union. It is responsible for the scientific evaluation, supervision, and safety monitoring of medicines in the EU. The European Commission also works to ensure that medical devices are safe and effective for use in the EU. In China, medical devices are regulated by the China National Medical Products Administration (NMPA) . It is important for manufacturers of medical devices to be familiar with the regulations and classifications set forth by the NMPA to ensure compliance and successfully bring their products to market in China.
Overview Of Approval Process For Medical Device In Major Markets
The FDA, European Commission & NMPA require manufacturers of medical devices to submit clinical data and other information to demonstrate that the devices are safe and effective before they can be sold in the respective markets. In addition, agencies have post-market surveillance programs in place to monitor the safety and performance of medical devices once they are on the market. There are several different types of medical devices, including:
- Diagnostic devices: These devices are used to diagnose medical conditions. Examples include blood glucose monitors, pregnancy tests, and CT scanners.
- Therapeutic devices: These devices are used to treat medical conditions. Examples include pacemakers, insulin pumps, and knee replacements.
- Monitoring devices: These devices are used to monitor patients’ vital signs or medical conditions. Examples include heart rate monitors, blood pressure monitors, and respiratory monitors.
- Rehabilitation devices: These devices are used to help patients recover from injuries or surgeries. Examples include crutches, wheelchairs, and prosthetics.
- In vitro diagnostic devices: These devices are used to test samples in a laboratory setting. Examples include blood glucose monitors, pregnancy tests, and cholesterol tests.
The FDA, European Commission & NMPA have strict guidelines for the approval and regulation of medical devices. To be approved for sale in the United States, EU, or China, A medical device must go through a thorough review process.
Medical Device Approval Process For The United States
As per FDA device classification, Class I medical devices which fall under the low risks category do not require premarket approval from FDA. However, the device must be registered on the FDA website and must comply with Labelling requirements. Class II and some class I medical devices which fall under the moderate risk category required FDA clearance in terms of premarket notification 510(K). Class III medical devices with High risk must complete the premarket approval process (PMA) which includes clinical study data of safety and effectiveness. The Novel devices which have low or moderate risk without a predicate are required to go with the De novo application process.
Medical Device Approval Process For Europe
A foreign manufacturer needs to appoint a local representative. The sponsor should determine, which category the device belongs to, If the device belongs to class I, non-sterile, and non-measuring, then a QMS is not formally required. However, a PMS procedure is required though not audited by a Notified Body (NB). The manufacturer must obtain a CE mark, which indicates that the device meets all relevant EU regulatory requirements. To obtain a CE mark, the manufacturer must complete a conformity assessment procedure, which typically includes a technical file review and a clinical evaluation. For those devices that belong to other classes, Quality Management System (QMS) is required, and most companies apply for ISO 13485 standards to achieve QMS compliance. The applicant shall prepare a technical file and demonstrate compliance. In the case of the class III device, a dossier has to be compiled. The QMS and the technical file (dossier in case of class III device) shall be audited by a notified body. For class I, non-sterile, and non-measuring, there is an audit or technical file required. Prepare a declaration of conformity.
Detailed overview of Medical Device Regulations in Europe.
Medical Device Approval Process For China
Class I – registration dossier – no testing reports Class II – full registration dossier and technical review Class III – full registration dossier and technical review The technical dossier that manufacturers are required to submit must contain thorough technical information, clinical data, and quality documentation. All Class II and III devices must be tested locally in China, though the NMPA may accept part of your previous test results. Depending on your device type, different testing criteria apply.
How Can RegDesk Help?
RegDesk is an AI-powered Regulatory Information Management System (RIMS) designed to simplify global compliance for medical device companies. With regulatory intelligence covering 120+ markets, RegDesk helps you prepare and publish global submissions, manage standards, conduct impact assessments, and stay ahead of regulatory changes all from a single, centralized platform. Expanding into new markets has never been easier.
