
Ireland medical device regulations are unified with other European Union countries and follow the same EU MDR and IVDR regulation. However, each country has their own Regulatory Authority (RA). In the case of Ireland, such RA is HPRA.

Overview
All medical devices in Ireland are regulated by Health Products Regulatory Authority (HPRA). However, HPRA (formerly known as Irish Medicines Board – IMB) is not responsible for medical device regulations alone. As of 2014, it is also responsible for regulating such areas as:
- cosmetic products
- clinical trials
- tissues and cells
- blood and blood components
- controlled drugs
- veterinary medicines
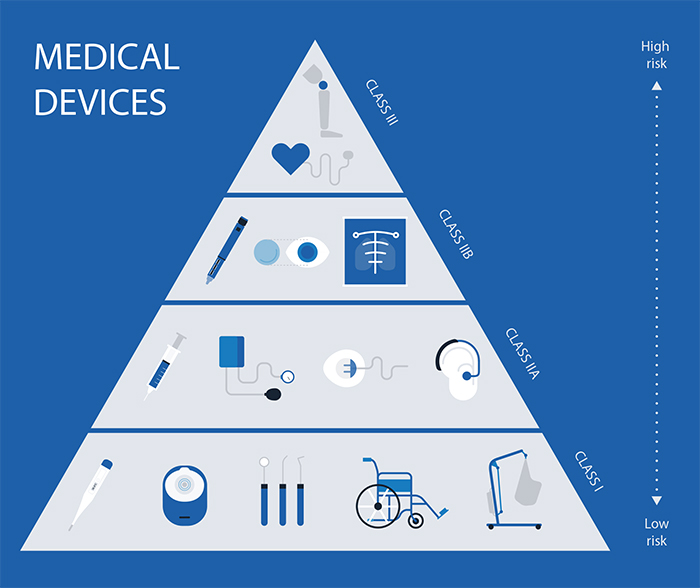
As medical devices in Ireland follow regulations established with all of the EU member states in mind, the classification system is the same as well. In Ireland, as well as all other EU member states, medical devices are classified into four different classes: Class I, Class IIa, Class IIb, and Class III.
Ireland Medical device registration process
In order to start marketing a medical devices in Ireland, manufacturers must first get them CE-marked. A CE mark is a sign of conformity with the European Union regulations that allows for the device to be marketed in any of the EU member states. It is enough to have a device successfully registered in Ireland, or any other EU country for the matter, and you will be able to freely market it anywhere on the EU territory.
Before starting the marketing process of medical devices, manufacturers should also look into a Quality Management System. One of the most popular choices is the ISO 13485 certification. It is important to remember, that this particular certification is only valid for three years, while all other EU licences are valid for five years.
The timeframe of the registration process depends mostly on the class of the device. Class 3 medical device registration can take even several months, while for Class I devices it should take no longer than 4 to 6 weeks. A Class 3 medical device is a device associated with the highest risk, so it is understandable why Regulatory Authorities have to be very thorough during the examination before allowing them to be marketed.
It is also important to remember that according to the current EU regulations, it is required from foreign manufacturers to appoint an Authorized Representative (AR) who will assist with the registration process. Foreign manufacturers, by definition, are all manufacturers that are registered outside of the European Union.




